

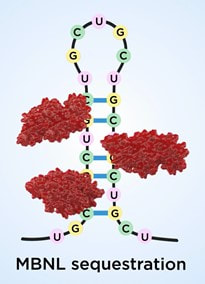
La Distrofia Miotónica tipo 1 es un trastorno genético heredable, causado por una alteración del gen DMPK (Dystrophia Myotonica Protein Kinase), ubicado en el cromosoma 19, concretamente la región 19q13.32. La enfermedad se debe a la presencia de un número elevado de repeticiones CTG en la secuencia 3’UTR, hacia el final de su secuencia. Al trascribirse este gen, se produce un ARN mensajero alterado, excesivamente largo, que genera estructuras secundarias, horquillas. Estos largos ARNm portadores de largas repeticiones CTG forman agregados en el núcleo y se acumulan de forma progresiva, hasta el punto de ser visibles al microscopio en músculo, corazón y neuronas. Estos agregados, llamados foci, son una seña de identidad de la DM1 y funcionan como una trampa para proteínas como las de la familia MBNL (Muscleblind like), que quedan secuestradas en estas estructuras; como consecuencia, se pierde su función en la célula.
|

|

Aunque hay otras proteínas cuyo metabolismo es alterado por las horquillas de repeticiones CTG del ARNm expandido (ej: las proteínas CELF ven su actividad aumentada), se ha observado en ratones mutantes para Mbnl1, que su sola falta de función provoca un fenotipo afín a lo que se observa en músculo de pacientes DM1, lo que convierte MBNL1 en un excelente objetivo terapéutico para la enfermedad. Las proteínas MBNL y CELF tienen un importante papel en los procesos de splicing alternativo que se dan en el procesamiento de genes, y que son necesarios para generar las distintas variantes de una proteína propias de cada tejido o estado del desarrollo (por ejemplo, hay variantes propias del embrión que no se producen en el adulto, y viceversa). La presencia de MBNL también regula un conjunto de microARNs que contribuyen a explicar las disfunciones que se observan en la célula de pacientes DM1, tal es el caso de miR-23b y miR-218.
La disfunción de proteínas encargadas de regular el patrón de splincing de otros muchos genes y mantener un adecuado balance de microARNs, tiene un profundo impacto en el funcionamiento de la célula y explica que la DM1 sea una enfermedad compleja y multisistémica. Como cabe esperar, los órganos en los que la DM1 se presenta con mayor severidad coincide con aquellos en los que el gen DMPK se expresa a mayores niveles, como es el caso del tejido muscular, ya que sus transcritos funcionan como ARN tóxico para la célula.
La disfunción de proteínas encargadas de regular el patrón de splincing de otros muchos genes y mantener un adecuado balance de microARNs, tiene un profundo impacto en el funcionamiento de la célula y explica que la DM1 sea una enfermedad compleja y multisistémica. Como cabe esperar, los órganos en los que la DM1 se presenta con mayor severidad coincide con aquellos en los que el gen DMPK se expresa a mayores niveles, como es el caso del tejido muscular, ya que sus transcritos funcionan como ARN tóxico para la célula.
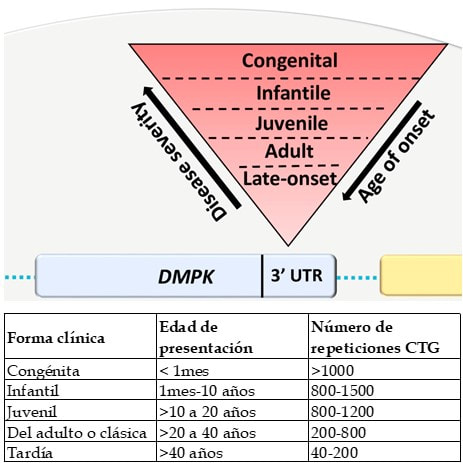
La herencia de este trastorno es dominante; es decir, es suficiente heredar una sola copia de DMPK expandido (del padre o de la madre) para desarrollar la enfermedad. Su naturaleza hace que también sea una enfermedad degenerativa, es decir, el ARN tóxico se va a cumulando en las células y sus efectos van apareciendo paulatinamente. La enfermedad se caracteriza por un amplio espectro clínico y diversos mecanismos moleculares subyacentes, lo que la convierte en uno de los trastornos genéticos más complejos que se conocen. El número de repeticiones CTG en el gen DMPK se correlaciona con la severidad de la enfermedad y el momento vital en el que se presenta la enfermedad en un paciente. Pese a ser una de las enfermedades neuromusculares más frecuentes, la variabilidad y progresividad con la que la DM1 se presenta clínicamente, hace que sea una afección habitualmente infradiagnosticada, lo que hace necesaria la concienciación sobre la enfermedad tanto entre profesionales de la salud como entre el público general.
La DM1 puede manifestarse en cualquier etapa de la vida, pudiéndose definir 5 formas de presentación, desde el tipo congénito a la forma de aparición tardía 10. La edad promedio de inicio de la clínica de la distrofia miotónica de Steinert es de 26,1 ± 13,2 años. El síntoma de comienzo más frecuente es el agarrotamiento de las manos (38,3 %) debido a la miotonía (contracción muscular con dificultad para la relajación) que limita o impide soltar objetos cogidos con la mano; el dolor se describe como manifestación inicial en el 3% de los casos. A veces pasa desapercibida, empeora con el frío y mejora al calentar el músculo. La debilidad y atrofia musculares son de predominio distal e intensidad variable, y se instauran progresivamente en los músculos faciales, masticatorios, del cuello, flexores de los dedos, antebrazos y flexores de los pies. El período de tiempo medio necesario para alcanzar el diagnóstico correcto es de 7,3 ± 8,2 años.
Diagnóstico de la DM1.
La función del médico de atención primaria, desde el punto de vista diagnóstico de la DM1, consiste en sospechar la existencia de una miopatía. La evaluación del paciente con debilidad muscular requiere una anamnesis y exploración física cuidadosas, a fin de diferenciar la debilidad muscular de otros síntomas (astenia, fatiga, dolor, rigidez o pérdida de fuerza de origen no muscular). La distribución de la debilidad muscular puede contribuir a diferenciar trastornos neurológicos y estrictamente miopáticos. Es habitual que los pacientes presenten niveles elevados de CPK (creatina-(fosfo)-kinasa) aislados, sin otra clínica acompañante. Cualquier proceso con lesión muscular puede elevar el nivel de CPK, pero elevaciones por encima de 5 veces su valor normal, indican claramente la existencia de miopatía. En las determinaciones rutinarias pueden encontrarse hipertransaminasemias asintomáticas (elevación de GOT y GPT), que se deben complementar con una determinación de CPK para saber son de origen muscular o hepático. Ante la sospecha de miopatía, conviene precisar con exactitud el tipo de miopatía de que se trata. La biopsia muscular, si bien no proporciona información relevante para el diagnóstico, puede mostrar en un estudio histológico gran cantidad de núcleos centrales, aglomeraciones de núcleos picnóticos, masas sarcoplásmicas, existencia de fibras en anillo, y atrofia selectiva de fibras tipo 1.
El diagnóstico de confirmación de la DM1 es competencia del neurólogo y se basa, junto con la exploración clínica, en el estudio electrofisiológico (electromiografía [EMG], para detectar descargas miotónicas típicas) y en el estudio genético. La Triple-repeat Primed-PCR (TP-PCR) es una técnica barata que detecta si el número de repeticiones CTG está en el rango normal o patológico, pero no permite cuantificarlas (detecta hasta 180 repeticiones). Se han demostrado unas tasas de prevalencia superiores al doble cuando se aplican técnicas de diagnóstico molecular en comparación con el diagnóstico basado exclusivamente en criterios clínicos6,7. Por lo tanto, el estudio genético molecular no se limita a la confirmación diagnóstica, sino que puede permitir la identificación de enfermos asintomáticos o con patología leve.
Como la mayoría de las miopatías tienen una base genética, el paciente puede transmitir la enfermedad a su descendencia y el fenotipo de los hijos puede no ser el mismo que el del padre o la madre. El asesoramiento genético sólo es practicable con un diagnóstico de la miopatía lo más exacto posible.
Anticipación Clínica en la DM1
Las evidencias indican que las secuencias CTG expandidas son genéticamente inestables y que existe una tendencia a aumentar el número de repeticiones en las células, en un fenómeno conocido como anticipación, del que se conocen pocos detalles. La inestabilidad provoca errores en la división celular, de modo que cada individuo es un mosaico, y distintos tejidos pueden presentar distinto número de repeticiones dentro del mismo paciente (el número de CTG en músculo esquelético suele ser entre 2 y 13 veces el encontrado en leucocitos). Esto afecta igualmente a las células reproductoras, de forma que la enfermedad se ve sometida a amplificación de una generación a la siguiente con, por lo general, un aumento de la gravedad de los síntomas.
Incluso en personas con 37-49 repeticiones CTG (llamado estado pre-mutacional) que no suelen presentar síntomas de DM1, estas repeticiones podrían verse expandidas, pudiendo afectar a futuras generaciones; esto explica que los hijos que heredan la mutación suelen presentar formas más graves que sus padres. El riesgo de tener un hijo con DM1 congénita es mayor cuanto mayor es la expansión materna, especialmente por encima de 300 CTG, pero esta posibilidad existe siempre que una mujer pasa la mutación a su descendencia, aun cuando su expansión no sea muy grande. Incluso una mujer asintomática o con una forma mínima de la enfermedad puede tener un hijo con la variante congénita.
Más raro es que se produzcan contracciones en la longitud del fragmento CTG que se transmite. Este fenómeno se da con más frecuencia cuando el transmisor es un varón con fragmentos grandes, por encima de 500 CTG.
La DM1 puede manifestarse en cualquier etapa de la vida, pudiéndose definir 5 formas de presentación, desde el tipo congénito a la forma de aparición tardía 10. La edad promedio de inicio de la clínica de la distrofia miotónica de Steinert es de 26,1 ± 13,2 años. El síntoma de comienzo más frecuente es el agarrotamiento de las manos (38,3 %) debido a la miotonía (contracción muscular con dificultad para la relajación) que limita o impide soltar objetos cogidos con la mano; el dolor se describe como manifestación inicial en el 3% de los casos. A veces pasa desapercibida, empeora con el frío y mejora al calentar el músculo. La debilidad y atrofia musculares son de predominio distal e intensidad variable, y se instauran progresivamente en los músculos faciales, masticatorios, del cuello, flexores de los dedos, antebrazos y flexores de los pies. El período de tiempo medio necesario para alcanzar el diagnóstico correcto es de 7,3 ± 8,2 años.
Diagnóstico de la DM1.
La función del médico de atención primaria, desde el punto de vista diagnóstico de la DM1, consiste en sospechar la existencia de una miopatía. La evaluación del paciente con debilidad muscular requiere una anamnesis y exploración física cuidadosas, a fin de diferenciar la debilidad muscular de otros síntomas (astenia, fatiga, dolor, rigidez o pérdida de fuerza de origen no muscular). La distribución de la debilidad muscular puede contribuir a diferenciar trastornos neurológicos y estrictamente miopáticos. Es habitual que los pacientes presenten niveles elevados de CPK (creatina-(fosfo)-kinasa) aislados, sin otra clínica acompañante. Cualquier proceso con lesión muscular puede elevar el nivel de CPK, pero elevaciones por encima de 5 veces su valor normal, indican claramente la existencia de miopatía. En las determinaciones rutinarias pueden encontrarse hipertransaminasemias asintomáticas (elevación de GOT y GPT), que se deben complementar con una determinación de CPK para saber son de origen muscular o hepático. Ante la sospecha de miopatía, conviene precisar con exactitud el tipo de miopatía de que se trata. La biopsia muscular, si bien no proporciona información relevante para el diagnóstico, puede mostrar en un estudio histológico gran cantidad de núcleos centrales, aglomeraciones de núcleos picnóticos, masas sarcoplásmicas, existencia de fibras en anillo, y atrofia selectiva de fibras tipo 1.
El diagnóstico de confirmación de la DM1 es competencia del neurólogo y se basa, junto con la exploración clínica, en el estudio electrofisiológico (electromiografía [EMG], para detectar descargas miotónicas típicas) y en el estudio genético. La Triple-repeat Primed-PCR (TP-PCR) es una técnica barata que detecta si el número de repeticiones CTG está en el rango normal o patológico, pero no permite cuantificarlas (detecta hasta 180 repeticiones). Se han demostrado unas tasas de prevalencia superiores al doble cuando se aplican técnicas de diagnóstico molecular en comparación con el diagnóstico basado exclusivamente en criterios clínicos6,7. Por lo tanto, el estudio genético molecular no se limita a la confirmación diagnóstica, sino que puede permitir la identificación de enfermos asintomáticos o con patología leve.
Como la mayoría de las miopatías tienen una base genética, el paciente puede transmitir la enfermedad a su descendencia y el fenotipo de los hijos puede no ser el mismo que el del padre o la madre. El asesoramiento genético sólo es practicable con un diagnóstico de la miopatía lo más exacto posible.
Anticipación Clínica en la DM1
Las evidencias indican que las secuencias CTG expandidas son genéticamente inestables y que existe una tendencia a aumentar el número de repeticiones en las células, en un fenómeno conocido como anticipación, del que se conocen pocos detalles. La inestabilidad provoca errores en la división celular, de modo que cada individuo es un mosaico, y distintos tejidos pueden presentar distinto número de repeticiones dentro del mismo paciente (el número de CTG en músculo esquelético suele ser entre 2 y 13 veces el encontrado en leucocitos). Esto afecta igualmente a las células reproductoras, de forma que la enfermedad se ve sometida a amplificación de una generación a la siguiente con, por lo general, un aumento de la gravedad de los síntomas.
Incluso en personas con 37-49 repeticiones CTG (llamado estado pre-mutacional) que no suelen presentar síntomas de DM1, estas repeticiones podrían verse expandidas, pudiendo afectar a futuras generaciones; esto explica que los hijos que heredan la mutación suelen presentar formas más graves que sus padres. El riesgo de tener un hijo con DM1 congénita es mayor cuanto mayor es la expansión materna, especialmente por encima de 300 CTG, pero esta posibilidad existe siempre que una mujer pasa la mutación a su descendencia, aun cuando su expansión no sea muy grande. Incluso una mujer asintomática o con una forma mínima de la enfermedad puede tener un hijo con la variante congénita.
Más raro es que se produzcan contracciones en la longitud del fragmento CTG que se transmite. Este fenómeno se da con más frecuencia cuando el transmisor es un varón con fragmentos grandes, por encima de 500 CTG.